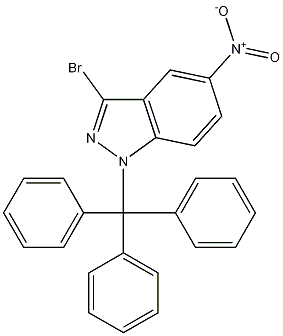
3-bromo-5-nitro-1-trityl-1H-indazole
- Product Name3-bromo-5-nitro-1-trityl-1H-indazole
- CAS942189-39-1
- CBNumberCB32499663
- MFC26H18BrN3O2
- MW484.34
- MOL File942189-39-1.mol
- MSDS FileSDS
Chemical Properties
| Boiling point | 637.5±55.0 °C(Predicted) |
| Density | 1.39±0.1 g/cm3(Predicted) |
| storage temp. | under inert gas (nitrogen or Argon) at 2-8°C |
| pka | -2.57±0.50(Predicted) |
3-bromo-5-nitro-1-trityl-1H-indazole Price
| Product number | Packaging | Price | Product description | Buy |
|---|---|---|---|---|
| Biosynth Carbosynth FB142080 | 25mg | $53 | 3-Bromo-5-nitro-1-trityl-1H-indazole |
Buy |
| Biosynth Carbosynth FB142080 | 50mg | $93 | 3-Bromo-5-nitro-1-trityl-1H-indazole |
Buy |
| AK Scientific 2353DP | 100mg | $105 | 3-Bromo-5-nitro-1-trityl-1H-indazole |
Buy |
| AK Scientific 2353DP | 250mg | $131 | 3-Bromo-5-nitro-1-trityl-1H-indazole |
Buy |
| Biosynth Carbosynth FB142080 | 100mg | $162 | 3-Bromo-5-nitro-1-trityl-1H-indazole |
Buy |
3-bromo-5-nitro-1-trityl-1H-indazole Chemical Properties,Usage,Production
Synthesis
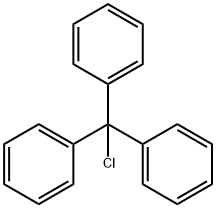
76-83-5
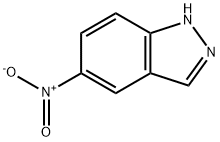
5401-94-5
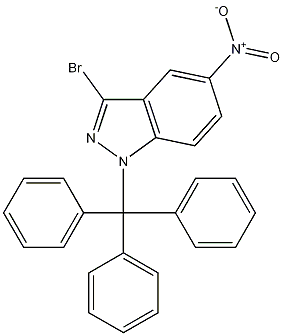
942189-39-1
The general procedure for the synthesis of 3-bromo-5-nitro-1-trityl-1H-indazole using triphenylchloromethane and 5-nitroindazole as starting materials was as follows: firstly, the compound formulation was divided into three intermediate formulations, A, B and C, followed by the coupling reaction of the intermediates. All intermediates were used as starting materials from commercially available compounds. Compound 5 was prepared by the reaction of bromo-4-cyanobenzene with methylhydrazine under acidic conditions to give hydrazinyl sialic acid ester in moderate yield.2 Subsequently, bromophenyl-N-methyltriazole intermediates were obtained by a two-step reaction with formic acid.3 A tetrahydropyridine ring was introduced by a Suzuki coupling reaction of commercially available Boc-protected tetrahydropyridine-boronic acid ester to form a tricyclic system.4 Next, the intermediates were separated into three intermediate preparations by the deprotected 4 with chloroacetyl chloride, chloroacetamide 5 was obtained in excellent yield.5 Starting from the commercially available 6, the pyrrolidine nucleus 10a was obtained in good yield after 5 steps.The reaction was carried out using thionyl chloride to give thiomethyl olefin 7.8 was obtained by cycloaddition reaction (2+3) followed by removal of the benzyl protecting group to give 9.Acid splitting was carried out in methanol using L-tartaric acid and filtration gave the pure ( S) enantiomer 9. After protection as a Boc derivative and methyl ester hydrolysis, 10 was obtained in 50% overall yield. Compound 17 was prepared from commercially available indazole 11. The bromination of indazole 11 at the 3-position afforded 12 in excellent yield without chromatographic separation.The bromo compound 12 was chromatographically separated to give nitroindazole 16 after Suzuki coupling of the bromo compound 12 with 14.The reduction of 16 afforded aniline 17 as an oil in quantitative yield without chromatographic separation. The final coupling of the intermediate was carried out by coupling 17 with 10a to give 18 in good yield.After removal of the Boc and triphenylmethyl protecting groups, the final coupling reaction with 5 afforded (S)-N-(3-(6-isopropoxypyridin-3-yl)-1H-indazol-5-yl)-1-(2-(2-(4-(4-(1-methyl-1H-1,2,4-triazol-3-yl)phenyl )-3,6-dihydropyridin-1(2H)-yl)-2-oxoethyl)-3-(methylthio)pyrrolidine-3-carboxamide followed by chromatographic separation. Final purification was accomplished by crystallization from methanol/diethyl ether. This synthetic route successfully prepared (S)-N-(3-(6-isopropoxypyridin-3-yl)-1H-indazol-5-yl)-1-(2-(4-(4-(1-methyl-1H-1,2,4-triazol-3-yl)phenyl)-3,6-dihydropyridin-1(2H)-yl)-2-oxoethyl)-3-(methylthio)pyrrolidine-3-carboxamide free base ( Compound I).
References
[1] Patent: WO2016/100152, 2016, A1. Location in patent: Page/Page column 8; 10[2] Patent: WO2016/100147, 2016, A1. Location in patent: Page/Page column 5; 8
[3] Patent: WO2017/40362, 2017, A1. Location in patent: Page/Page column 4; 6
Preparation Products And Raw materials
3-bromo-5-nitro-1-trityl-1H-indazole Supplier
| Supplier | Tel | Country | ProdList | Advantage | |
|---|---|---|---|---|---|
| +86-(0)57185586718; +8613336195806 |
sales@capot.com | China | 29640 | 60 | |
| +86-27-50766799 +8618062016861 |
contact@labnetwork.com | China | 19987 | 58 | |
| +undefined18143011203 | info@chemextension.com | China | 42056 | 58 | |
| 571-89925085 | sales@amadischem.com | China | 131957 | 58 | |
| 13526569071 | sales@leadmedpharm.com | China | 7891 | 64 | |
| 021-54306202 13764082696 |
info@hanhongsci.com | China | 42934 | 64 | |
| 021-20432229 | 2412110556@qq.com | China | 6017 | 55 | |
| 400-711-5280 86-571-81025280 | sales@molcore.com | China | 9967 | 58 | |
| 025-58851090 15955137747 |
sales01@aikonchem.com | China | 15949 | 58 | |
| 021-58432009 400-005-6266 |
marketing@energy-chemical.com | China | 44918 | 58 |
The What'sApp is temporarily not supported in mainland China