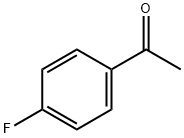
Fluorination Effects of 4-Fluoroacetophenone
4-Fluoroacetophenone is a versatile aromatic ketone that serves as a crucial intermediate in organic synthesis and pharmaceutical applications. With its unique fluorine substitution, this compound enhances the reactivity and selectivity in various chemical reactions, making it an invaluable asset for researchers and industry professionals. It is commonly utilized in the synthesis of pharmaceuticals, agrochemicals, and fine chemicals, where its ability to modify biological activity is particularly beneficial.

Fluorination effects probed in 4-fluoroacetophenone
Due to its small radius and high electronegativity, the introduction of the fluorine atom into known compound scaffolds is one of the efficient strategies to optimize chemical properties or biological activities, such as reducing the cytotoxicity of the drug, avoiding protein denaturation and strengthening the role of drug molecules. At the molecular level, these changes upon fluorination might also arise from the subtle interplay among various non-covalent interactions (NCIs) at play. After perfluorination of the methyl group of AP, the substitution makes the water oxygen link with the aromatic CB–H, forming a cyclic CB–H⋯OW and OW–H⋯OC HBs in the trifluoroacetophenone (TFAP)–water complex. According to the previous studies, 4-fluorotoluene has almost the same barrier as toluene, suggesting that the 4-fluorination seems to have smallest influence on electronic structure of toluene with respect to 2- and 3-fluorinations. Nevertheless, we suspect that the 4-fluorination may still affect the topologies of NCIs. To disclose how the fluorination affects the methyl internal rotation, geometries and interaction preference of AP, the rotational spectra of 4-fluoroacetophenone (4FAP) and its monohydrate were investigated by pulsed jet FTMW spectroscopy combined with quantum chemical calculations. Rotational spectra of the 4-Fluoroacetophenone monomer and its monohydrate were measured using a coaxially oriented beam-resonator arrangement (COBRA) pulsed-jet FTMW spectrometer (covering 2–20 GHz), operating with the FTMW++ aquisition and analysis program.[1]
The structure of the 4FAP monomer optimized at the MP2/6-311++G(d,p) level is shown, where the principal inertial axes and heavy atomic labels are also given. The theoretical values of spectroscopic parameters including equilibrium rotational (Ae, Be, Ce) and centrifugal distortion constants (DJ, DK, DJK, d1 and d2) as well as the electric dipole moment components (μa, μb, and μc) are reported. Six stationary points were obtained on the MP2/6-311++G(d,p) potential energy surface of 4-Fluoroacetophenone–H2O. Their corresponding theoretical values of spectroscopic parameters are listed. In the two most stable isomers I and II, water serves as a proton donor forming an OW–H⋯OC HB. Compared with two most stable isomers of AP–H2O, the 4-fluorine substitution results in a smaller energy difference between isomer I and isomer II, and the global minimum changes to isomer II when the BSSE correction is included. However, the value of the V3 barrier to methyl internal rotation for isomer II increases noticeably from that of the 4FAP monomer which is similar to 4-hydroxy-2-butanone–H2O. The results indicate that the different binding site of the water molecule can tune the electronic structure of 4-Fluoroacetophenone. Assuming that most vibrational population has been frozen to the respective ground states by molecular collision during the early stages of the supersonic expansion, information on conformational population of isomers I and II of 4-Fluoroacetophenone–H2O in the jet expansion can be derived from the relative intensities of rotational transitions.
By using the high resolution pulsed-jet Fourier transform microwave technique combined with theoretical calculations, the pure rotational spectra of 4-Fluoroacetophenone and its monohydrate have been investigated. Only one conformer of 4FAP has been observed and the barrier to the methyl internal rotation was determined to be 7.25(1) kJ mol−1. Rotational spectra of the normal species and all 13C isotopologues were observed in natural abundance, which allowed the accurate determination of the structure of the heavy atom skeleton. For 4-Fluoroacetophenone–H2O, rotational spectra of two isomers and their water isotopic species were observed in the pulsed jet with a derived population ratio of about NI/NII ≈ 7/1. Both measured isomers are stabilized by OW–H⋯OC and C–H⋯OW HBs. Splittings arising from methyl internal rotation in both isomers were observed as A-/E-symmetry species doublets. The estimated V3 barrier of isomer I (V3 = 7.10(3) kJ mol−1) is close to that of the 4-Fluoroacetophenone monomer (V3 = 7.25(1) kJ mol−1), while the value of V3 the barrier of isomer II (V3 = 8.886(6) kJ mol−1) is higher than that of the 4-Fluoroacetophenone monomer. It shows that the electronic structure of 4FAP can be tuned by the binding site of the water molecule. The analyses of non-covalent interactions pointed out that the 4-fluorination decreases the electronic density of the π system and further enhances the capacity of benzene hydrogen acting as proton donor.
References
[1]Wang, Xiujuan et al. “Fluorination effects probed in 4-fluoroacetophenone and its monohydrate.” Physical chemistry chemical physics : PCCP vol. 25,37 25450-25457. 27 Sep. 2023, doi:10.1039/d3cp01578e
Lastest Price from 4-Fluoroacetophenone manufacturers
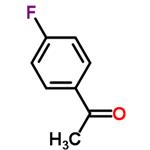
US $0.00-0.00/kg2025-11-20
- CAS:
- 403-42-9
- Min. Order:
- 1kg
- Purity:
- 98%
- Supply Ability:
- 100tons

US $10.00/ASSAYS2025-08-29
- CAS:
- 403-42-9
- Min. Order:
- 1ASSAYS
- Purity:
- 99%
- Supply Ability:
- 1 ton